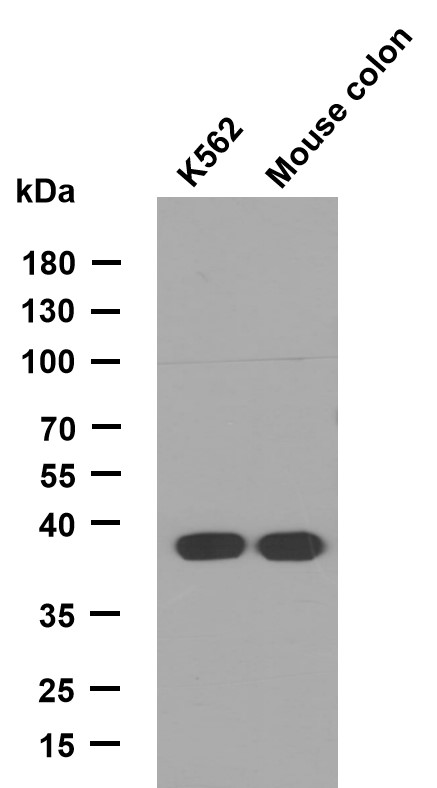
FHL1 rabbit pAb
- Catalog No.:YT7722
- Applications:WB
- Reactivity:Human;Mouse;Rat
- Target:
- FHL1
- Fields:
- >>JAK-STAT signaling pathway
- Gene Name:
- FHL1 SLIM1
- Protein Name:
- FHL1
- Human Gene Id:
- 2273
- Human Swiss Prot No:
- Q13642
- Mouse Gene Id:
- 14199
- Mouse Swiss Prot No:
- P97447
- Rat Gene Id:
- 25177
- Rat Swiss Prot No:
- Q9WUH4
- Immunogen:
- Synthesized peptide derived from human FHL1 AA range: 66-116
- Specificity:
- This antibody detects endogenous levels of FHL1 at Human/Mouse/Rat
- Formulation:
- Liquid in PBS containing 50% glycerol, 0.5% BSA and 0.02% sodium azide.
- Source:
- Polyclonal, Rabbit,IgG
- Dilution:
- WB 1:500-2000
- Purification:
- The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen.
- Concentration:
- 1 mg/ml
- Storage Stability:
- -15°C to -25°C/1 year(Do not lower than -25°C)
- Molecular Weight(Da):
- 36kD
- Background:
- This gene encodes a member of the four-and-a-half-LIM-only protein family. Family members contain two highly conserved, tandemly arranged, zinc finger domains with four highly conserved cysteines binding a zinc atom in each zinc finger. Expression of these family members occurs in a cell- and tissue-specific mode and these proteins are involved in many cellular processes. Mutations in this gene have been found in patients with Emery-Dreifuss muscular dystrophy. Multiple alternately spliced transcript variants which encode different protein isoforms have been described.[provided by RefSeq, Nov 2009],
- Function:
- developmental stage:Elevated levels during postnatal muscle growth.,disease:Defects in FHL1 are the cause of X-linked childhood-onset reducing body myopathy (RBM) [MIM:300718]. This disorder is allelic to severe early-onset reducing body myopathy (RBM) [MIM:300717].,disease:Defects in FHL1 are the cause of X-linked dominant scapuloperoneal myopathy [MIM:300695]. Scapuloperoneal syndrome (SPS) was initially described more than 120 years ago by Jules Broussard as 'une forme hereditaire d'atrophie musculaire progressive' beginning in the lower legs and affecting the shoulder region earlier and more severely than distal arm. The etiology of this condition remains unclear.,disease:Defects in FHL1 are the cause of X-linked myopathy with postural muscle atrophy (XMPMA) [MIM:300696]. Myopathies are inherited muscle disorders characterized by weakness and atrophy of voluntary skeletal muscle, and
- Subcellular Location:
- [Isoform 1]: Cytoplasm.; [Isoform 3]: Cytoplasm. Nucleus.; [Isoform 2]: Nucleus. Cytoplasm, cytosol. Predominantly nuclear in myoblasts but is cytosolic in differentiated myotubes.
- Expression:
- Isoform 1 is highly expressed in skeletal muscle and to a lesser extent in heart, placenta, ovary, prostate, testis, small intestine, colon and spleen. Expression is barely detectable in brain, lung, liver, kidney, pancreas, thymus and peripheral blood leukocytes. Isoform 2 is expressed in brain, skeletal muscle and to a lesser extent in heart, colon, prostate and small intestine. Isoform 3 is expressed in testis, heart and skeletal muscle.
- June 19-2018
- WESTERN IMMUNOBLOTTING PROTOCOL
- June 19-2018
- IMMUNOHISTOCHEMISTRY-PARAFFIN PROTOCOL
- June 19-2018
- IMMUNOFLUORESCENCE PROTOCOL
- September 08-2020
- FLOW-CYTOMEYRT-PROTOCOL
- May 20-2022
- Cell-Based ELISA│解您多样本WB检测之困扰
- July 13-2018
- CELL-BASED-ELISA-PROTOCOL-FOR-ACETYL-PROTEIN
- July 13-2018
- CELL-BASED-ELISA-PROTOCOL-FOR-PHOSPHO-PROTEIN
- July 13-2018
- Antibody-FAQs
- Products Images

- Western blot analysis of lysates from PC-12 cells, primary antibody was diluted at 1:1000, 4°over night