ND5 Polyclonal Antibody
- Catalog No.:YT3001
- Applications:WB;ELISA
- Reactivity:Human;Rat;Mouse;
- Target:
- ND5
- Fields:
- >>Oxidative phosphorylation;>>Metabolic pathways;>>Thermogenesis;>>Retrograde endocannabinoid signaling;>>Alzheimer disease;>>Parkinson disease;>>Amyotrophic lateral sclerosis;>>Huntington disease;>>Prion disease;>>Pathways of neurodegeneration - multiple diseases;>>Chemical carcinogenesis - reactive oxygen species;>>Diabetic cardiomyopathy
- Gene Name:
- MT-ND5
- Protein Name:
- NADH-ubiquinone oxidoreductase chain 5
- Human Gene Id:
- 4540
- Human Swiss Prot No:
- P03915
- Mouse Swiss Prot No:
- P03921
- Immunogen:
- The antiserum was produced against synthesized peptide derived from human MT-ND5. AA range:328-377
- Specificity:
- ND5 Polyclonal Antibody detects endogenous levels of ND5 protein.
- Formulation:
- Liquid in PBS containing 50% glycerol, 0.5% BSA and 0.02% sodium azide.
- Source:
- Polyclonal, Rabbit,IgG
- Dilution:
- WB 1:500 - 1:2000. ELISA: 1:10000. Not yet tested in other applications.
- Purification:
- The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen.
- Concentration:
- 1 mg/ml
- Storage Stability:
- -15°C to -25°C/1 year(Do not lower than -25°C)
- Other Name:
- MT-ND5;MTND5;NADH5;ND5;NADH-ubiquinone oxidoreductase chain 5;NADH dehydrogenase subunit 5
- Observed Band(KD):
- 70kD
- Background:
- catalytic activity:NADH + ubiquinone = NAD(+) + ubiquinol.,disease:Defects in MT-ND5 are a cause of complex I mitochondrial respiratory chain deficiency [MIM:252010]. Complex I (NADH-ubiquinone oxidoreductase), the largest complex of the mitochondrial respiratory chain, contains more than 40 subunits. It is embedded in the inner mitochondrial membrane and is partly protruding in the matrix. Complex I deficiency is the most common cause of mitochondrial disorders. It represents largely one-third of all cases of respiratory chain deficiency and is responsible for a variety of clinical symptoms, ranging from neurological disorders to cardiomyopathy, liver failure, and myopathy.,disease:Defects in MT-ND5 are a cause of Leber hereditary optic neuropathy (LHON) [MIM:535000]. LHON is a maternally inherited disease resulting in acute or subacute loss of central vision, due to optic nerve dysfunction. Cardiac conduction defects and neurological defects have also been described in some patients. LHON results from primary mitochondrial DNA mutations affecting the respiratory chain complexes.,disease:Defects in MT-ND5 are a cause of Leigh syndrome (LS) [MIM:256000]. LS is a severe neurological disorder characterized by bilaterally symmetrical necrotic lesions in subcortical brain regions.,disease:Defects in MT-ND5 are a cause of mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes syndrome (MELAS) [MIM:540000]. MELAS is a genetically heterogenious disorder, characterized by episodic vomiting, seizures, and recurrent cerebral insults resembling strokes and causing hemiparesis, hemianopsia, or cortical blindness.,disease:Defects in MT-ND5 are associated with features of myoclonic epilepsy associated with ragged-red fibers (MERRF) [MIM:545000]. MERRF is a mitochondrial encephalomyopathy characterized by myoclonic seizures. The prevalence in the general population of Europe has been estimated at 0.9 in 100'000 individuals, but the disease seems to be more common in the USA. Patients usually present during adolescence or early adulthood with myoclonic epilepsy, sometimes with neurosensory deafness, optic atrophy, short stature or peripheral neuropathy.,function:Core subunit of the mitochondrial membrane respiratory chain NADH dehydrogenase (Complex I) that is believed to belong to the minimal assembly required for catalysis. Complex I functions in the transfer of electrons from NADH to the respiratory chain. The immediate electron acceptor for the enzyme is believed to be ubiquinone.,similarity:Belongs to the complex I subunit 5 family.,
- Function:
- catalytic activity:NADH + ubiquinone = NAD(+) + ubiquinol.,disease:Defects in MT-ND5 are a cause of complex I mitochondrial respiratory chain deficiency [MIM:252010]. Complex I (NADH-ubiquinone oxidoreductase), the largest complex of the mitochondrial respiratory chain, contains more than 40 subunits. It is embedded in the inner mitochondrial membrane and is partly protruding in the matrix. Complex I deficiency is the most common cause of mitochondrial disorders. It represents largely one-third of all cases of respiratory chain deficiency and is responsible for a variety of clinical symptoms, ranging from neurological disorders to cardiomyopathy, liver failure, and myopathy.,disease:Defects in MT-ND5 are a cause of Leber hereditary optic neuropathy (LHON) [MIM:535000]. LHON is a maternally inherited disease resulting in acute or subacute loss of central vision, due to optic nerve dysfunc
- Subcellular Location:
- Mitochondrion inner membrane ; Multi-pass membrane protein .
- Expression:
- Blood,Bone fossil,Bones,Breast cancer,Distant normal tissue,Glioma,
- June 19-2018
- WESTERN IMMUNOBLOTTING PROTOCOL
- June 19-2018
- IMMUNOHISTOCHEMISTRY-PARAFFIN PROTOCOL
- June 19-2018
- IMMUNOFLUORESCENCE PROTOCOL
- September 08-2020
- FLOW-CYTOMEYRT-PROTOCOL
- May 20-2022
- Cell-Based ELISA│解您多样本WB检测之困扰
- July 13-2018
- CELL-BASED-ELISA-PROTOCOL-FOR-ACETYL-PROTEIN
- July 13-2018
- CELL-BASED-ELISA-PROTOCOL-FOR-PHOSPHO-PROTEIN
- July 13-2018
- Antibody-FAQs
- Products Images

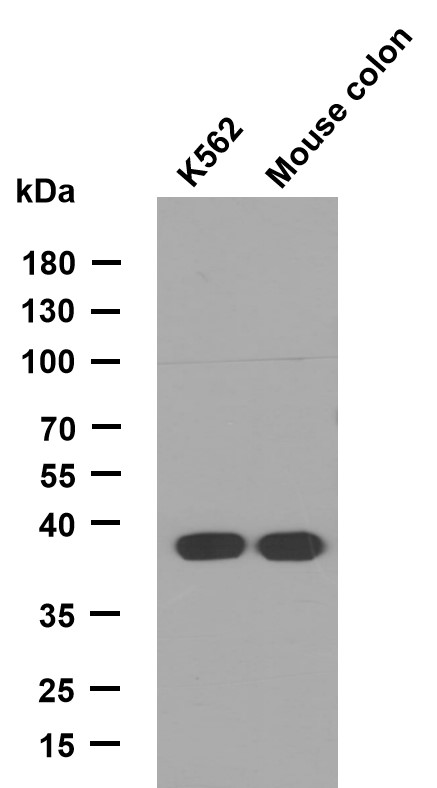
- Western Blot analysis of various cells using ND5 Polyclonal Antibody diluted at 1:1000
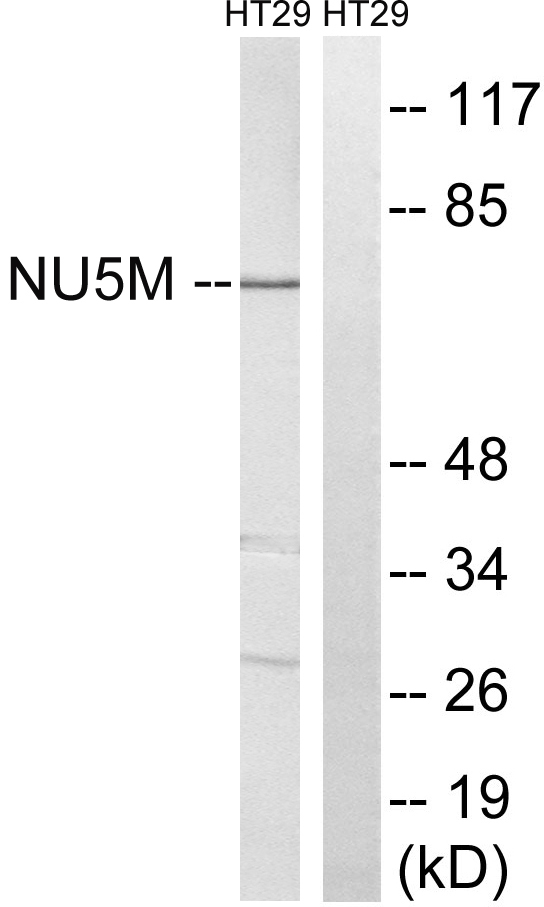
- Western blot analysis of lysates from HT-29 cells, using MT-ND5 Antibody. The lane on the right is blocked with the synthesized peptide.

- Western blot analysis of the lysates from Jurkat cells using MT-ND5 antibody.