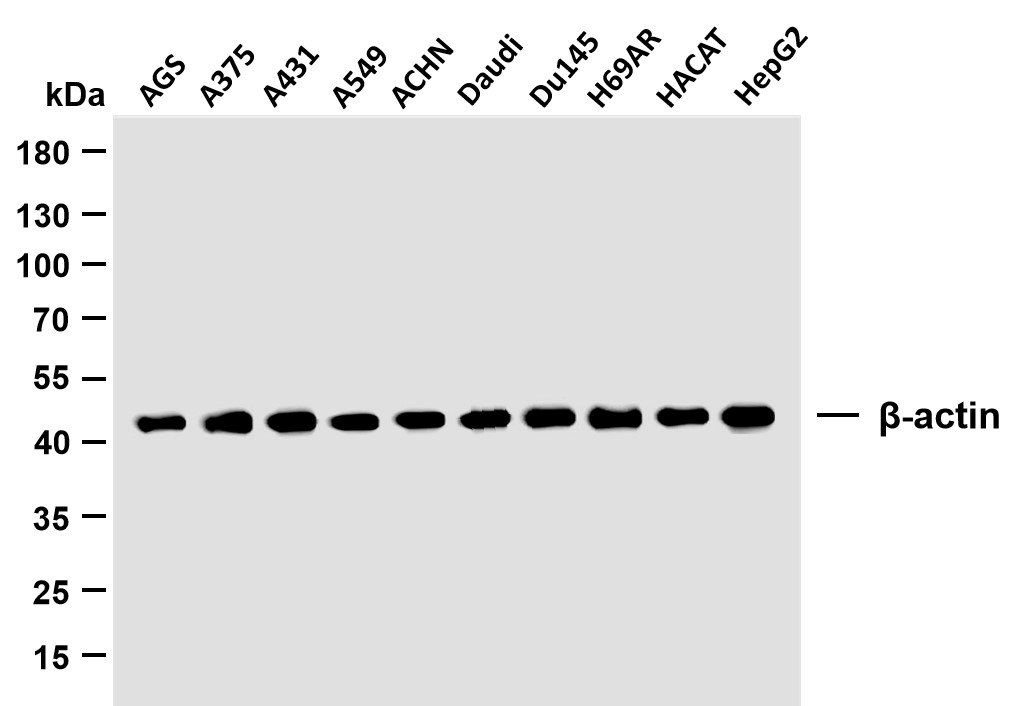
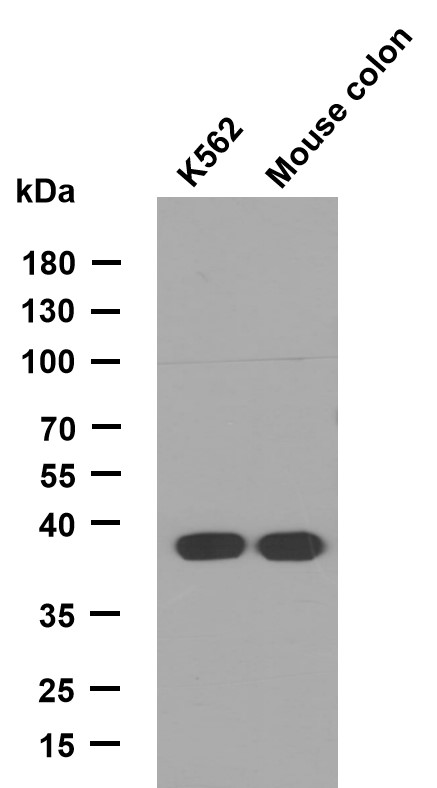
Glucuronidase β Polyclonal Antibody
- Catalog No.:YT1920
- Applications:WB;IHC;IF;ELISA
- Reactivity:Human;Mouse;Rat
- Target:
- Glucuronidase β
- Fields:
- >>Pentose and glucuronate interconversions;>>Ascorbate and aldarate metabolism;>>Glycosaminoglycan degradation;>>Porphyrin metabolism;>>Drug metabolism - other enzymes;>>Metabolic pathways;>>Biosynthesis of cofactors;>>Lysosome
- Gene Name:
- GUSB
- Protein Name:
- Beta-glucuronidase
- Human Gene Id:
- 2990
- Human Swiss Prot No:
- P08236
- Mouse Gene Id:
- 110006
- Mouse Swiss Prot No:
- P12265
- Rat Gene Id:
- 24434
- Rat Swiss Prot No:
- P06760
- Immunogen:
- The antiserum was produced against synthesized peptide derived from human GUSB. AA range:321-370
- Specificity:
- Glucuronidase β Polyclonal Antibody detects endogenous levels of Glucuronidase β protein.
- Formulation:
- Liquid in PBS containing 50% glycerol, 0.5% BSA and 0.02% sodium azide.
- Source:
- Polyclonal, Rabbit,IgG
- Dilution:
- IHC: 100-300.WB 1:500 - 1:2000. ELISA: 1:10000.. IF 1:50-200
- Purification:
- The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen.
- Concentration:
- 1 mg/ml
- Storage Stability:
- -15°C to -25°C/1 year(Do not lower than -25°C)
- Other Name:
- GUSB;Beta-glucuronidase;Beta-G1
- Observed Band(KD):
- 78kD
- Background:
- This gene encodes a hydrolase that degrades glycosaminoglycans, including heparan sulfate, dermatan sulfate, and chondroitin-4,6-sulfate. The enzyme forms a homotetramer that is localized to the lysosome. Mutations in this gene result in mucopolysaccharidosis type VII. Alternative splicing results in multiple transcript variants. There are many pseudogenes of this locus in the human genome.[provided by RefSeq, May 2014],
- Function:
- catalytic activity:A beta-D-glucuronoside + H(2)O = D-glucuronate + an alcohol.,disease:Defects in GUSB are the cause of mucopolysaccharidosis type 7 (MPS7) [MIM:253220]; also known as Sly syndrome. MPS7 is an autosomal recessive lysosomal storage disease characterized by inability to degrade glucuronic acid-containing glycosaminoglycans. The phenotype is highly variable, ranging from severe lethal hydrops fetalis to mild forms with survival into adulthood. Most patients with the intermediate phenotype show hepatomegaly, skeletal anomalies, coarse facies, and variable degrees of mental impairment.,disease:Mucopolysaccharidosis type 7 is associated with non-immune hydrops fetalis [MIM:236750]. Hydrops fetalis is a generalized edema of the fetus with fluid accumulation in the body cavities.,enzyme regulation:Inhibited by L-aspartic acid.,function:Plays an important role in the degradation
- Subcellular Location:
- Lysosome.
- Expression:
- Colon,Fibroblast,Liver,Placenta,Plasma,
- June 19-2018
- WESTERN IMMUNOBLOTTING PROTOCOL
- June 19-2018
- IMMUNOHISTOCHEMISTRY-PARAFFIN PROTOCOL
- June 19-2018
- IMMUNOFLUORESCENCE PROTOCOL
- September 08-2020
- FLOW-CYTOMEYRT-PROTOCOL
- May 20-2022
- Cell-Based ELISA│解您多样本WB检测之困扰
- July 13-2018
- CELL-BASED-ELISA-PROTOCOL-FOR-ACETYL-PROTEIN
- July 13-2018
- CELL-BASED-ELISA-PROTOCOL-FOR-PHOSPHO-PROTEIN
- July 13-2018
- Antibody-FAQs
- Products Images
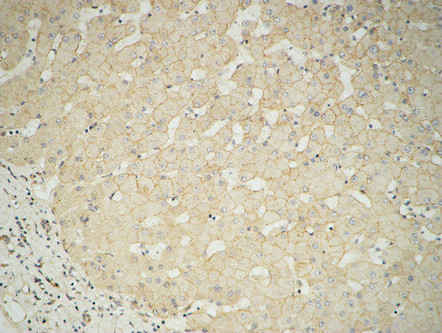
- Immunohistochemical analysis of paraffin-embedded Human Liver. 1, Antibody was diluted at 1:100(4° overnight). 2, High-pressure and temperature EDTA, pH8.0 was used for antigen retrieval. 3,Secondary antibody was diluted at 1:200(room temperature, 30min).

- Western blot analysis of the lysates from COLO205 cells using GUSB antibody.