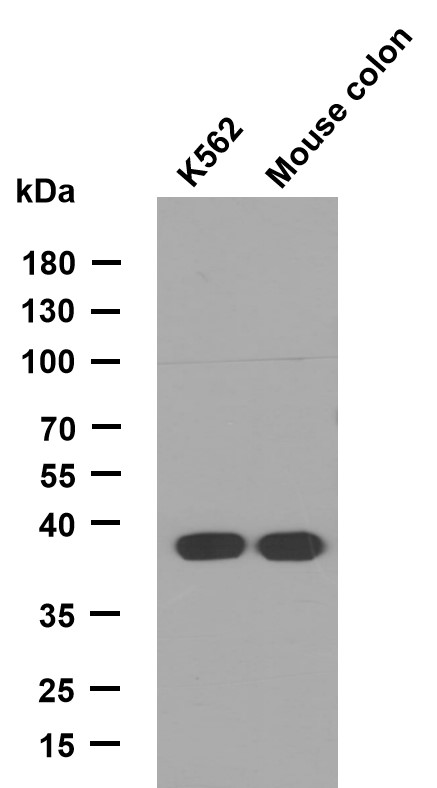
GAS3 Polyclonal Antibody
- Catalog No.:YT1851
- Applications:WB;IHC;IF;ELISA
- Reactivity:Human;Mouse;Rat
- Target:
- GAS3
- Gene Name:
- PMP22
- Protein Name:
- Peripheral myelin protein 22
- Human Gene Id:
- 5376
- Human Swiss Prot No:
- Q01453
- Mouse Gene Id:
- 18858
- Mouse Swiss Prot No:
- P16646
- Rat Gene Id:
- 24660
- Rat Swiss Prot No:
- P25094
- Immunogen:
- The antiserum was produced against synthesized peptide derived from human PMP22. AA range:111-160
- Specificity:
- GAS3 Polyclonal Antibody detects endogenous levels of GAS3 protein.
- Formulation:
- Liquid in PBS containing 50% glycerol, 0.5% BSA and 0.02% sodium azide.
- Source:
- Polyclonal, Rabbit,IgG
- Dilution:
- WB 1:500 - 1:2000. IHC 1:100 - 1:300. ELISA: 1:40000.. IF 1:50-200
- Purification:
- The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen.
- Concentration:
- 1 mg/ml
- Storage Stability:
- -15°C to -25°C/1 year(Do not lower than -25°C)
- Other Name:
- PMP22;GAS3;Peripheral myelin protein 22;PMP-22;Growth arrest-specific protein 3;GAS-3
- Observed Band(KD):
- 22kD
- Background:
- This gene encodes an integral membrane protein that is a major component of myelin in the peripheral nervous system. Studies suggest two alternately used promoters drive tissue-specific expression. Various mutations of this gene are causes of Charcot-Marie-Tooth disease Type IA, Dejerine-Sottas syndrome, and hereditary neuropathy with liability to pressure palsies. Alternative splicing results in multiple transcript variants. [provided by RefSeq, Jul 2013],
- Function:
- disease:Defects in PMP22 are a cause of Dejerine-Sottas syndrome (DSS) [MIM:145900]; also known as Dejerine-Sottas neuropathy (DSN) or hereditary motor and sensory neuropathy III (HMSN3). DSS is a severe degenerating neuropathy of the demyelinating Charcot-Marie-Tooth disease category, with onset by age 2 years. DSS is characterized by motor and sensory neuropathy with very slow nerve conduction velocities, increased cerebrospinal fluid protein concentrations, hypertrophic nerve changes, delayed age of walking as well as areflexia. There are both autosomal dominant and autosomal recessive forms of Dejerine-Sottas syndrome.,disease:Defects in PMP22 are a cause of hereditary neuropathy with liability to pressure palsies (HNPP) [MIM:162500]; an autosomal dominant disorder characterized by transient episodes of decreased perception or peripheral nerve palsies after slight traction, compressi
- Subcellular Location:
- Cell membrane ; Multi-pass membrane protein .
- Expression:
- Fetal fibroblast,Kidney,Peripheral blood,Peripheral blood leukocyte,Spinal
- June 19-2018
- WESTERN IMMUNOBLOTTING PROTOCOL
- June 19-2018
- IMMUNOHISTOCHEMISTRY-PARAFFIN PROTOCOL
- June 19-2018
- IMMUNOFLUORESCENCE PROTOCOL
- September 08-2020
- FLOW-CYTOMEYRT-PROTOCOL
- May 20-2022
- Cell-Based ELISA│解您多样本WB检测之困扰
- July 13-2018
- CELL-BASED-ELISA-PROTOCOL-FOR-ACETYL-PROTEIN
- July 13-2018
- CELL-BASED-ELISA-PROTOCOL-FOR-PHOSPHO-PROTEIN
- July 13-2018
- Antibody-FAQs
- Products Images
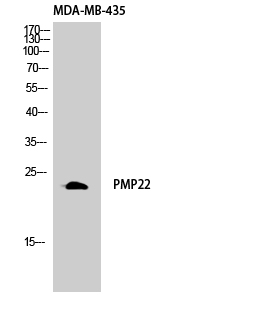
- Western Blot analysis of MDA-MB-435 cells using GAS3 Polyclonal Antibody diluted at 1:1000
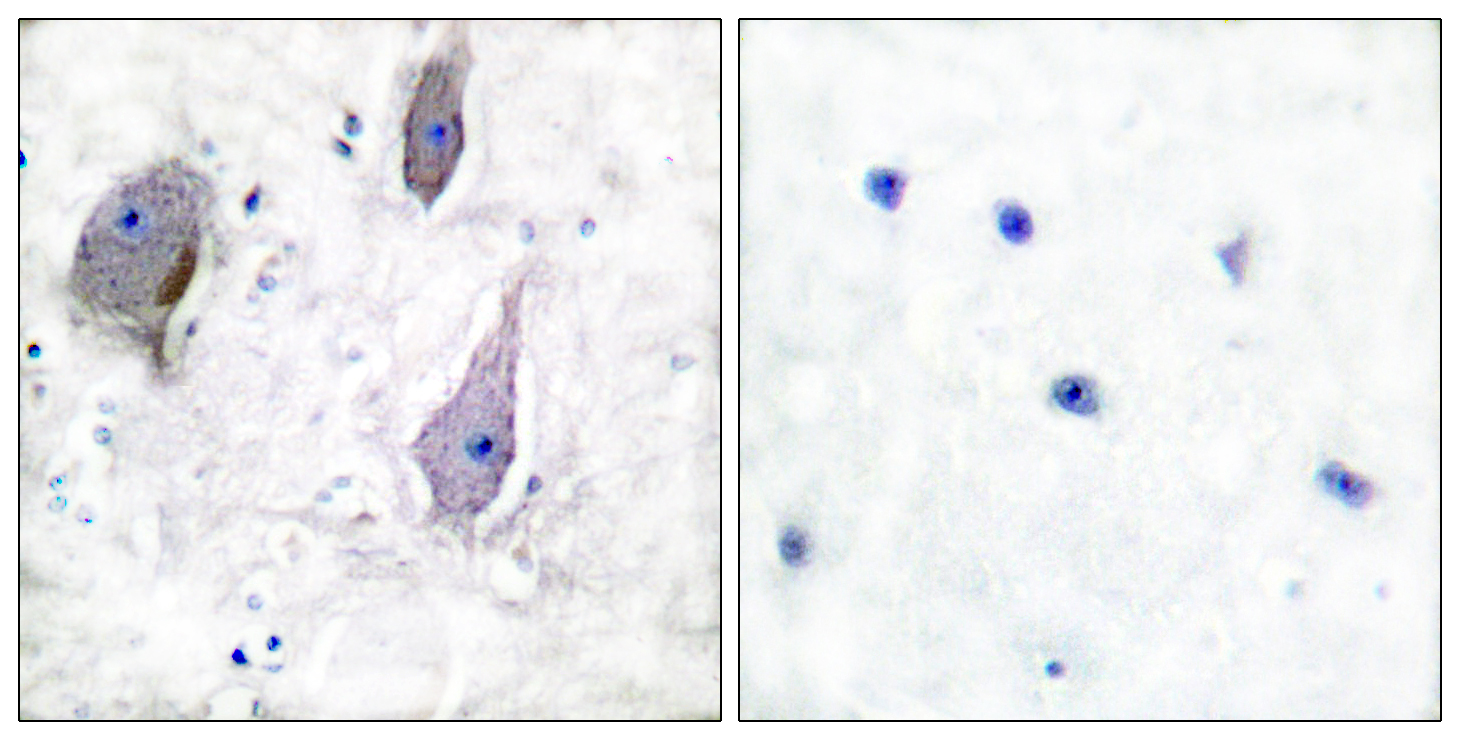
- Immunohistochemistry analysis of paraffin-embedded human brain tissue, using PMP22 Antibody. The picture on the right is blocked with the synthesized peptide.

- Western blot analysis of lysates from MDA-MB-435 cells, using PMP22 Antibody. The lane on the right is blocked with the synthesized peptide.