- Home
- About
- Promotions
-
Products
-
Elisa Kits
- |
-
Primary antibodies
- |
-
Secondary antibodies
- |
-
Proteins
- |
-
IHC reagents
- |
-
WB reagents
- PonceauS Staining Solution
- PBST Washing Buffer, 10X
- 1.5M Tris-HCl Buffer, pH8.8
- 1M Tris-HCl Buffer, pH6.8
- 10% SDS Solution
- Prestained Protein Marker
- TBST Washing Buffer, 10X
- SDS PAGE Loading Buffer, 5X
- Stripping Buffered Solution
- Tris Buffer, pH7.4, 10X
- Total Protein Extraction Kit
- Running Buffer, 10X
- Transfer Buffer, 10X
- 30% Acr-Bis(29:1) Solution
- Tris电泳液速溶颗粒
- PBS(1X, premixed powder)
- TBS(1X, premixed powder)
- 快速封闭液
- 转膜液速溶颗粒
- Chemical reagents
- News
- Distributor
- Resources
- Contact
- Home
- >
- Info
- >
- NPC1 rabbit pAb
- >
- Go Back
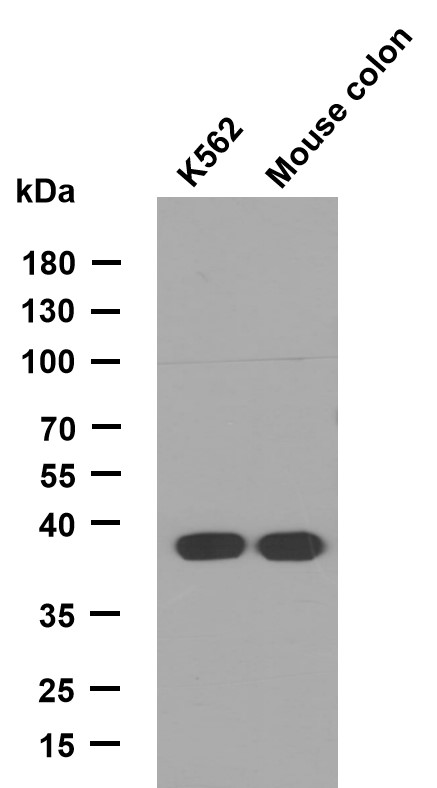
NPC1 rabbit pAb
- Catalog No.:YT7297
- Applications:WB
- Reactivity:Human;Mouse
- Fields:
- >>Lysosome;>>Cholesterol metabolism
- Immunogen:
- Synthesized peptide derived from human NPC1 AA range: 1073-1123
- Specificity:
- This antibody detects endogenous levels of NPC1 at Human/Mouse
- Formulation:
- Liquid in PBS containing 50% glycerol, 0.5% BSA and 0.02% sodium azide.
- Source:
- Polyclonal, Rabbit,IgG
- Purification:
- The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen.
- Storage Stability:
- -15°C to -25°C/1 year(Do not lower than -25°C)
- Molecular Weight(Da):
- 141kD
- Background:
- This gene encodes a large protein that resides in the limiting membrane of endosomes and lysosomes and mediates intracellular cholesterol trafficking via binding of cholesterol to its N-terminal domain. It is predicted to have a cytoplasmic C-terminus, 13 transmembrane domains, and 3 large loops in the lumen of the endosome - the last loop being at the N-terminus. This protein transports low-density lipoproteins to late endosomal/lysosomal compartments where they are hydrolized and released as free cholesterol. Defects in this gene cause Niemann-Pick type C disease, a rare autosomal recessive neurodegenerative disorder characterized by over accumulation of cholesterol and glycosphingolipids in late endosomal/lysosomal compartments.[provided by RefSeq, Aug 2009],
- Function:
- disease:Defects in NPC1 are the cause of Niemann-Pick disease type C1 (NPC1) [MIM:257220]. NPC1 is an autosomal recessive lipid storage disorder, which affects particularly the brain, liver and spleen, and which is characterized by lysosomal accumulation of low density lipoprotein derived cholesterol. Clinical features include variable hepatosplenomegaly and severe progressive neurological dysfunction such as ataxia, dystonia and dementia. The age of onset can vary from infancy to late adulthood.,disease:Defects in NPC1 are the cause of Niemann-Pick disease type D (NPD) [MIM:257220]; also known as Niemann-Pick disease without sphingomyelinase deficiency, or Nova Scotian type. Because of evidence from biochemical changes, lack of complementation, and linkage mapping to the same chromosome site, NPD and NPC1 are considered to be allelic disorders.,domain:A cysteine-rich N-terminal domain a
- Subcellular Location:
- Late endosome membrane ; Multi-pass membrane protein . Lysosome membrane ; Multi-pass membrane protein .

- Western blot analysis of lysates from KB cells, primary antibody was diluted at 1:1000, 4°over night