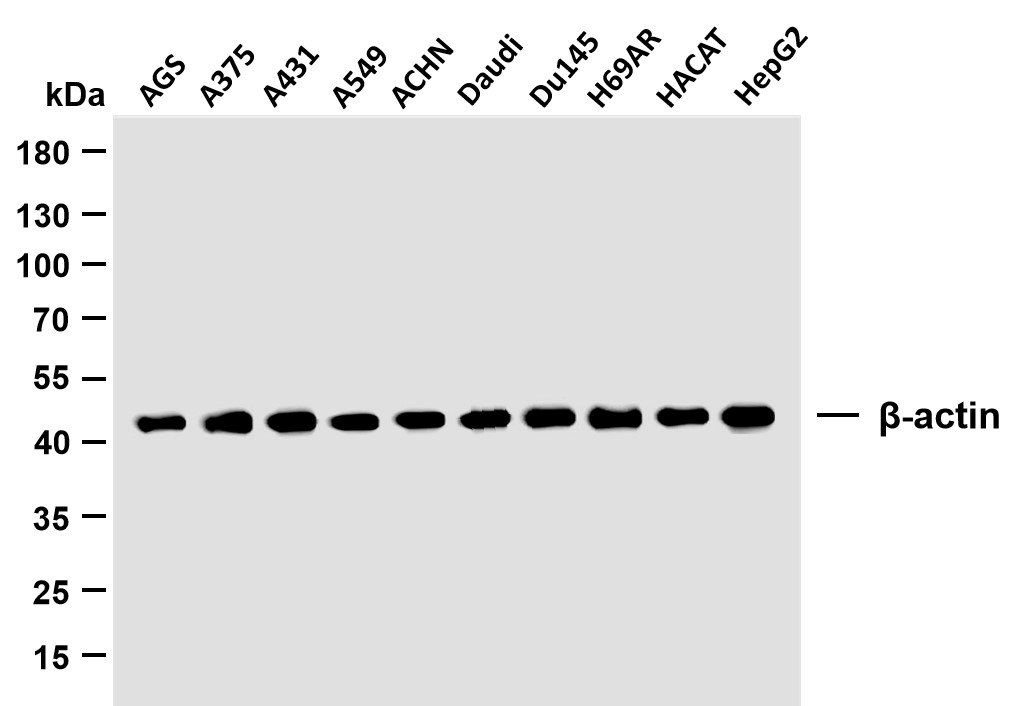
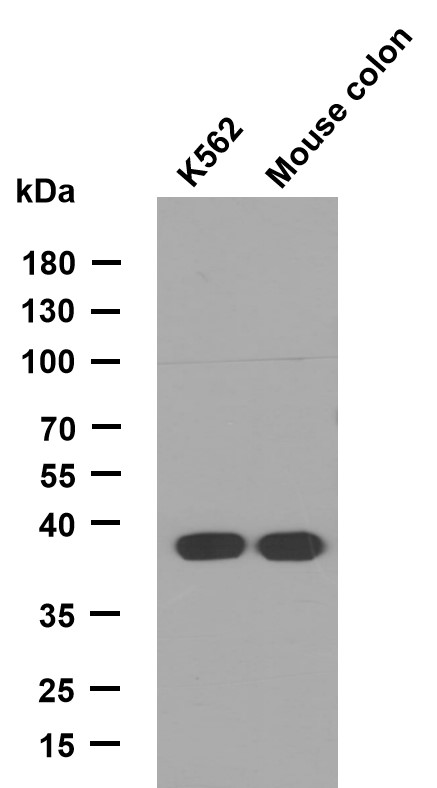
LIS1 rabbit pAb
- Catalog No.:YT6353
- Applications:WB;ELISA;IHC
- Reactivity:Human;Mouse;Rat
- Target:
- LIS1
- Fields:
- >>Ether lipid metabolism;>>Metabolic pathways
- Gene Name:
- PAFAH1B1 LIS1 MDCR MDS PAFAHA
- Protein Name:
- LIS1
- Human Gene Id:
- 5048
- Human Swiss Prot No:
- P43034
- Mouse Gene Id:
- 18472
- Mouse Swiss Prot No:
- P63005
- Rat Gene Id:
- 83572
- Rat Swiss Prot No:
- P63004
- Immunogen:
- Synthesized peptide derived from human LIS1
- Specificity:
- This antibody detects endogenous levels of LIS1 at Human/Mouse/Rat
- Formulation:
- Liquid in PBS containing 50% glycerol, 0.5% BSA and 0.02% sodium azide.
- Source:
- Polyclonal, Rabbit,IgG
- Dilution:
- WB 1:500-2000;IHC 1:50-300; ELISA 2000-20000
- Purification:
- The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen.
- Concentration:
- 1 mg/ml
- Storage Stability:
- -15°C to -25°C/1 year(Do not lower than -25°C)
- Molecular Weight(Da):
- 45kD
- Background:
- This locus was identified as encoding a gene that when mutated or lost caused the lissencephaly associated with Miller-Dieker lissencephaly syndrome. This gene encodes the non-catalytic alpha subunit of the intracellular Ib isoform of platelet-activating factor acteylhydrolase, a heterotrimeric enzyme that specifically catalyzes the removal of the acetyl group at the SN-2 position of platelet-activating factor (identified as 1-O-alkyl-2-acetyl-sn-glyceryl-3-phosphorylcholine). Two other isoforms of intracellular platelet-activating factor acetylhydrolase exist: one composed of multiple subunits, the other, a single subunit. In addition, a single-subunit isoform of this enzyme is found in serum. [provided by RefSeq, Apr 2009],
- Function:
- disease:Defects in PAFAH1B1 are a cause of Miller-Dieker lissencephaly syndrome (MDLS) [MIM:247200]. MDLS is a contiguous gene deletion syndrome of chromosome 17p13.3, characterized by classical lissencephaly and distinct facial features. Additional congenital malformations can be part of the condition.,disease:Defects in PAFAH1B1 are the cause of lissencephaly type 1 (LIS1) [MIM:607432]; also known as classic lissencephaly. LIS1 is characterized by agyria or pachgyria and disorganization of the clear neuronal lamination of normal six-layered cortex. The cortex is abnormally thick and poorly organized with 4 primitive layers. LIS1 is associated with enlarged and dysmorphic ventricles and often hypoplasia of the corpus callosum.,disease:Defects in PAFAH1B1 are the cause of subcortical band heterotopia (SBH) [MIM:607432]. SBH is a mild brain malformation of the lissencephaly spectrum. It i
- Subcellular Location:
- Cytoplasm, cytoskeleton. Cytoplasm, cytoskeleton, microtubule organizing center, centrosome . Cytoplasm, cytoskeleton, spindle . Nucleus membrane . Redistributes to axons during neuronal development. Also localizes to the microtubules of the manchette in elongating spermatids and to the meiotic spindle in spermatocytes (By similarity). Localizes to the plus end of microtubules and to the centrosome. May localize to the nuclear membrane. .
- Expression:
- Fairly ubiquitous expression in both the frontal and occipital areas of the brain.
- June 19-2018
- WESTERN IMMUNOBLOTTING PROTOCOL
- June 19-2018
- IMMUNOHISTOCHEMISTRY-PARAFFIN PROTOCOL
- June 19-2018
- IMMUNOFLUORESCENCE PROTOCOL
- September 08-2020
- FLOW-CYTOMEYRT-PROTOCOL
- May 20-2022
- Cell-Based ELISA│解您多样本WB检测之困扰
- July 13-2018
- CELL-BASED-ELISA-PROTOCOL-FOR-ACETYL-PROTEIN
- July 13-2018
- CELL-BASED-ELISA-PROTOCOL-FOR-PHOSPHO-PROTEIN
- July 13-2018
- Antibody-FAQs
- Products Images
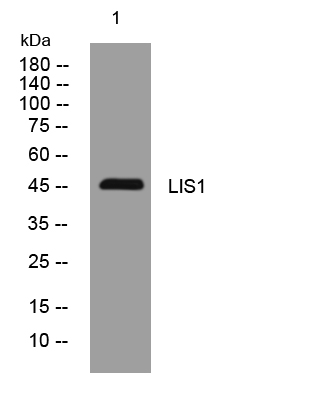
- Western blot analysis of lysates from KB cells, primary antibody was diluted at 1:1000, 4°over night
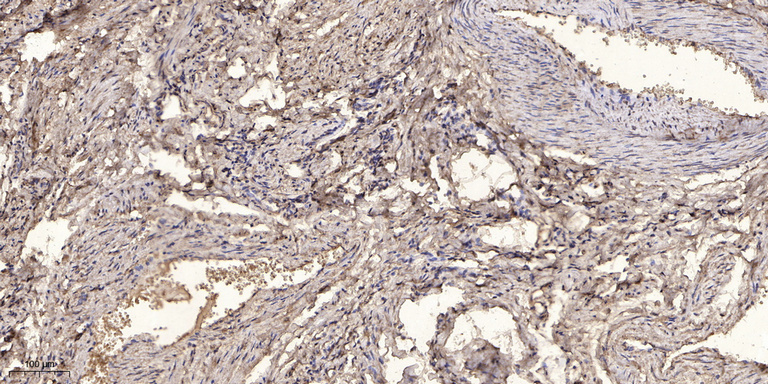
- Immunohistochemical analysis of paraffin-embedded human oophoroma. 1, Antibody was diluted at 1:200(4° overnight). 2, Tris-EDTA,pH9.0 was used for antigen retrieval. 3,Secondary antibody was diluted at 1:200(room temperature, 45min).