LPL Monoclonal Antibody
- Catalog No.:YM0420
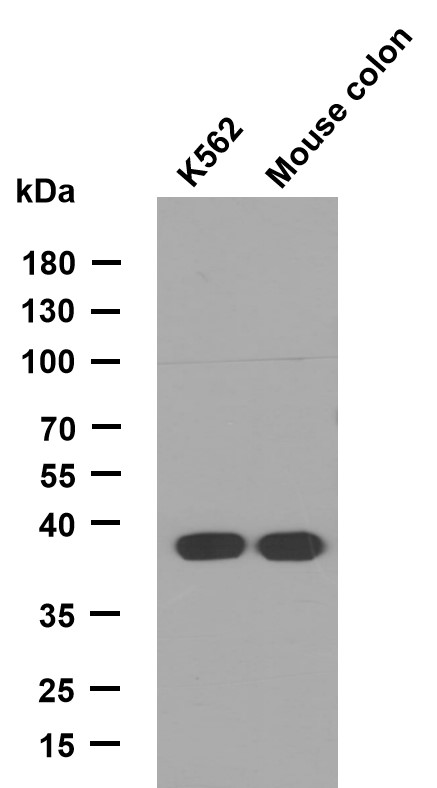
- Applications:WB;ELISA
- Reactivity:Human
- Target:
- LPL
- Fields:
- >>Glycerolipid metabolism;>>PPAR signaling pathway;>>Cholesterol metabolism;>>Alzheimer disease
- Gene Name:
- LPL
- Protein Name:
- Lipoprotein lipase
- Human Gene Id:
- 4023
- Human Swiss Prot No:
- P06858
- Mouse Swiss Prot No:
- P11152
- Immunogen:
- Purified recombinant fragment of LPL expressed in E. Coli.
- Specificity:
- LPL Monoclonal Antibody detects endogenous levels of LPL protein.
- Formulation:
- Liquid in PBS containing 50% glycerol, 0.5% BSA and 0.02% sodium azide.
- Source:
- Monoclonal, Mouse
- Dilution:
- WB 1:500 - 1:2000. ELISA: 1:10000. Not yet tested in other applications.
- Purification:
- Affinity purification
- Storage Stability:
- -15°C to -25°C/1 year(Do not lower than -25°C)
- Other Name:
- LPL;LIPD;Lipoprotein lipase;LPL
- Molecular Weight(Da):
- 53kD
- References:
- 1. Obesity (Silver Spring). 2008 Jan;16(1):199-201.
2. Hum Mutat. 2009 Jan;30(1):49-55.
- Background:
- lipoprotein lipase(LPL) Homo sapiens LPL encodes lipoprotein lipase, which is expressed in heart, muscle, and adipose tissue. LPL functions as a homodimer, and has the dual functions of triglyceride hydrolase and ligand/bridging factor for receptor-mediated lipoprotein uptake. Severe mutations that cause LPL deficiency result in type I hyperlipoproteinemia, while less extreme mutations in LPL are linked to many disorders of lipoprotein metabolism. [provided by RefSeq, Jul 2008],
- Function:
- catalytic activity:Triacylglycerol + H(2)O = diacylglycerol + a carboxylate.,disease:Defects in LPL are a cause of familial chylomicronemia [MIM:238600]; also known as hyperlipoproteinemia type I. Familial chylomicronemia is a recessive disorder usually manifesting in childhood. On a normal diet, patients often present with abdominal pain, hepatosplenomegaly, lipemia retinalis, eruptive xanthomata, and massive hypertriglyceridemia, sometimes complicated with acute pancreatitis.,disease:Defects in LPL are the cause of lipoprotein lipase deficiency (LPL deficiency) [MIM:238600]. LPL deficiency leads to hypertriglyceridemia.,function:The primary function of this lipase is the hydrolysis of triglycerides of circulating chylomicrons and very low density lipoproteins (VLDL). The enzyme functions in the presence of apolipoprotein C-2 on the luminal surface of vascular endothelium.,online inform
- Subcellular Location:
- Cell membrane ; Peripheral membrane protein ; Extracellular side . Secreted . Secreted, extracellular space, extracellular matrix . Newly synthesized LPL binds to cell surface heparan proteoglycans and is then released by heparanase. Subsequently, it becomes attached to heparan proteoglycan on endothelial cells (PubMed:27811232). Locates to the plasma membrane of microvilli of hepatocytes with triglyceride-rich lipoproteins (TRL). Some of the bound LPL is then internalized and located inside non-coated endocytic vesicles (By similarity). .
- Expression:
- Detected in blood plasma (PubMed:2340307, PubMed:11893776, PubMed:12641539). Detected in milk (at protein level) (PubMed:2340307).
- June 19-2018
- WESTERN IMMUNOBLOTTING PROTOCOL
- June 19-2018
- IMMUNOHISTOCHEMISTRY-PARAFFIN PROTOCOL
- June 19-2018
- IMMUNOFLUORESCENCE PROTOCOL
- September 08-2020
- FLOW-CYTOMEYRT-PROTOCOL
- May 20-2022
- Cell-Based ELISA│解您多样本WB检测之困扰
- July 13-2018
- CELL-BASED-ELISA-PROTOCOL-FOR-ACETYL-PROTEIN
- July 13-2018
- CELL-BASED-ELISA-PROTOCOL-FOR-PHOSPHO-PROTEIN
- July 13-2018
- Antibody-FAQs
- Products Images

- Western Blot analysis using LPL Monoclonal Antibody against HeLa cell lysate (1).