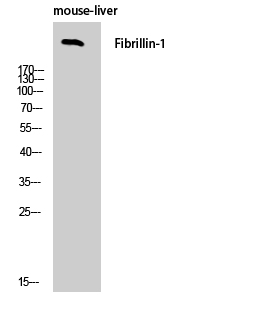
Total FBN1 Cell-Based Colorimetric ELISA Kit
- Catalog No.:KA3261C
- Applications:ELISA
- Reactivity:Human;Mouse;Rat
- Gene Name:
- FBN1
- Human Gene Id:
- 2200
- Human Swiss Prot No:
- P35555
- Mouse Swiss Prot No:
- Q61554
- Storage Stability:
- 2-8°C/6 months
- Other Name:
- Fibrillin-1
- Detection Method:
- Colorimetric
- Background:
- disease:Defects in FBN1 are a cause of isolated ectopia lentis (EL) [MIM:129600]. The symptoms of this autosomal dominant fibrillinopathy overlap with those of Marfan syndrome, with the exclusion of the skeletal and cardiovascular manifestations.,disease:Defects in FBN1 are a cause of Marfan syndrome (MFS) [MIM:154700]. MFS is an autosomal dominant disorder that affects the skeletal, ocular, and cardiovascular systems. A wide variety of skeletal abnormalities occurs with MFS, including scoliosis, chest wall deformity, tall stature, abnormal joint mobility. Ectopia lentis occurs in up to about 80% of MFS patients and is almost always bilateral. The leading cause of premature death in MFS patients is progressive dilation of the aortic root and ascending aorta, causing aortic incompetence and dissection. The majority of the more than 600 mutations in FBN1 currently known are point mutations, the rest are frameshifts and splice site mutations. Marfan syndrome has been suggested in at least 2 historical figures, Abraham Lincoln and Paganini.,disease:Defects in FBN1 are a cause of MASS syndrome [MIM:604308]. MASS syndrome is a heritable disorder of connective tissue characterized by involvement of the mitral valve, aorta, skeleton, and skin. MASS syndrome is closely resembling both the Marfan syndrome and the Barlow syndrome. However, no dislocation of the lenses or aneurysmal changes occur in the aorta, and the mitral valve prolapse is by no means invariable.,disease:Defects in FBN1 are a cause of Shprintzen-Goldberg craniosynostosis syndrome (SGS) [MIM:182212]. SGS is a very rare syndrome characterized by a marfanoid habitus, craniosynostosis, characteristic dysmorphic facial features, skeletal and cardiovascular abnormalities, mental retardation, developmental delay and learning disabilities.,disease:Defects in FBN1 are the cause of autosomal dominant Weill-Marchesani syndrome (WMS) [MIM:608328]. WMS is a rare connective tissue disorder characterized by short stature, brachydactyly, joint stiffness, and eye abnormalities including microspherophakia, ectopia lentis, severe myopia and glaucoma.,function:Fibrillins are structural components of 10-12 nm extracellular calcium-binding microfibrils, which occur either in association with elastin or in elastin-free bundles. Fibrillin-1-containing microfibrils provide long-term force bearing structural support.,online information:Fibrillin 1 mutation database,PTM:Forms intermolecular disulfide bonds either with other fibrillin-1 molecules or with other components of the microfibrils.,similarity:Belongs to the fibrillin family.,similarity:Contains 47 EGF-like domains.,similarity:Contains 9 TB (TGF-beta binding) domains.,subunit:Interacts with COL16A1.,
- Function:
- skeletal system development, urogenital system development, kidney development, heart development,
- Subcellular Location:
- Secreted . Fibrillin-1 and Asprosin chains are still linked together during the secretion from cells, but are subsequently separated by furin (PubMed:24982166). .; [Fibrillin-1]: Secreted, extracellular space, extracellular matrix .; [Asprosin]: Secreted . Secreted by white adipose tissue and circulates in the plasma. .
- June 19-2018
- WESTERN IMMUNOBLOTTING PROTOCOL
- June 19-2018
- IMMUNOHISTOCHEMISTRY-PARAFFIN PROTOCOL
- June 19-2018
- IMMUNOFLUORESCENCE PROTOCOL
- September 08-2020
- FLOW-CYTOMEYRT-PROTOCOL
- May 20-2022
- Cell-Based ELISA│解您多样本WB检测之困扰
- July 13-2018
- CELL-BASED-ELISA-PROTOCOL-FOR-ACETYL-PROTEIN
- July 13-2018
- CELL-BASED-ELISA-PROTOCOL-FOR-PHOSPHO-PROTEIN
- July 13-2018
- Antibody-FAQs