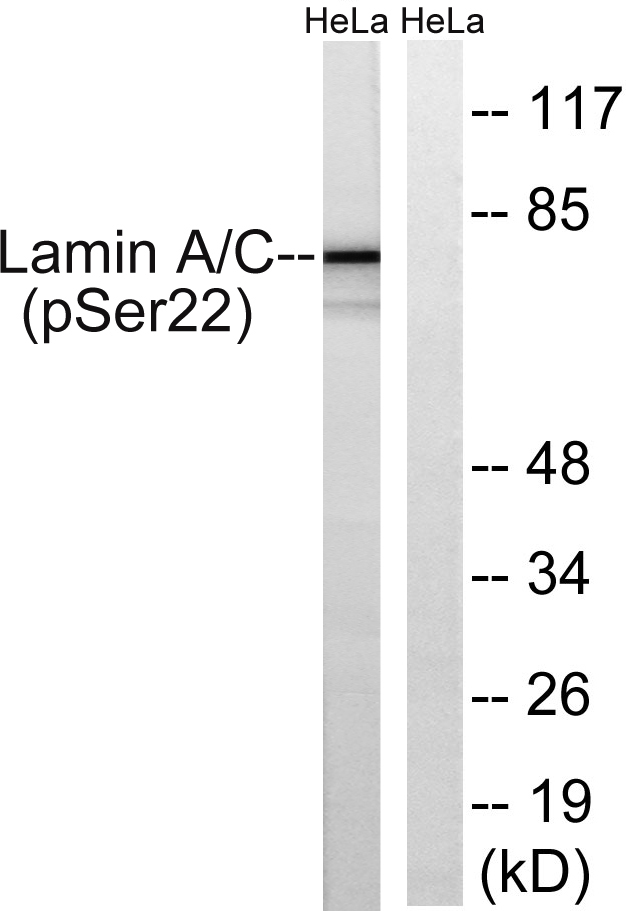
Total Lamin A/C Cell-Based Colorimetric ELISA Kit
- 货号:KA4158C
- 应用:ELISA
- 种属:Human;Mouse;Rat
- 基因名称:
- LMNA
- Human Gene Id:
- 4000
- Human Swiss Prot No:
- P02545
- Mouse Swiss Prot No:
- P48678
- Rat Swiss Prot No:
- P48679
- 储存:
- 2-8°C/6 months
- 其他名称:
- Prelamin-A/C [Cleaved into: Lamin-A/C (70 kDa lamin) (Renal carcinoma antigen NY-REN-32)]
- 检测方法:
- Colorimetric
- 背景:
- disease:Defects in LMNA are a cause of Emery-Dreifuss muscular dystrophy type 2 (EDMD2) [MIM:181350]. EDMD2 is an autosomal dominant disorder characterized by slowly progressive muscle wasting and weakness, early contractures of the elbows Achilles tendons and spine, and cardiomyopathy associated with cardiac conduction defects.,disease:Defects in LMNA are a cause of Emery-Dreifuss muscular dystrophy type 3 (EDMD3) [MIM:604929]. EDMD3 is an autosomal recessive disorder characterized by early contractures, muscle wasting and weakness and cardiomyopathy.,disease:Defects in LMNA are a cause of familial partial lipodystrophy type 2 (FPLD2) [MIM:151660]; also known as familial partial lipodystrophy Dunnigan type. FPLD2 is an autosomal dominant disorder characterized by marked loss of subcutaneous adipose tissue from the extremities and trunk but by excess fat deposition in the head and neck. Frequently associated with profound insulin resistance, dyslipidemia, and diabetes.,disease:Defects in LMNA are a cause of generalized lipoatrophy associated with diabetes, hepatic steatosis, hypertrophic cardiomyopathy and leukomelanodermic papules (LDHCP) [MIM:608056]. LDHCP is a disorder characterized by acquired generalized lipoatrophy with metabolic alterations, massive liver steatosis, distinctive cutaneous manifestations, and cardiac abnormalities involving both endocardium and myocardium.,disease:Defects in LMNA are a cause of lethal tight skin contracture syndrome [MIM:275210]; also called restrictive dermopathy (RD). Lethal tight skin contracture syndrome is a rare disorder mainly characterized by intrauterine growth retardation, tight and rigid skin with erosions, prominent superficial vasculature and epidermal hyperkeratosis, facial features (small mouth, small pinched nose and micrognathia), sparse/absent eyelashes and eyebrows, mineralization defects of the skull, thin dysplastic clavicles, pulmonary hypoplasia, multiple joint contractures and an early neonatal lethal course. Liveborn children usually die within the first week of life. The overall prevalence of consanguineous cases suggested an autosomal recessive inheritance.,disease:Defects in LMNA are a cause of tendinous calcinosis arthropathy and progeroid features (TCAPF) [MIM:611618]. This disorder consists of an autosomal recessive arthropathy syndrome affecting predominantly the distal femora and proximal tibia in the knees with tendinous calcifications, associated with progeroide appearance, such as pinched nose and micrognathia, cataract, alopecia, generalized lipodystrophy and sclerodermatous skin.,disease:Defects in LMNA are a cause of Werner syndrome (WRN) [MIM:277700]. WRN is an autosomal, recessively inherited, segmental progeroid syndrome, in which multiple aspects (or segments) of aging phenotypes seem to be entailed. The features of Werner syndrome are scleroderma-like skin changes, especially in the extremities, cataract, subcutaneous calcification, premature arteriosclerosis, diabetes mellitus, and a wizened and prematurely aged facies.,disease:Defects in LMNA are the cause of cardiomyopathy dilated type 1A (CMD1A) [MIM:115200]. Dilated cardiomyopathy is a disorder characterized by ventricular dilation and impaired systolic function, resulting in congestive heart failure and arrhythmia. Patients are at risk of premature death.,disease:Defects in LMNA are the cause of Charcot-Marie-Tooth disease type 2B1 (CMT2B1) [MIM:605588]. CMT2B1 is a form of Charcot-Marie-Tooth disease, the most common inherited disorder of the peripheral nervous system. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathy or CMT1, and primary peripheral axonal neuropathy or CMT2. Neuropathies of the CMT2 group are characterized by signs of axonal regeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. CMT2B1 inheritance is autosomal recessive.,disease:Defects in LMNA are the cause of heart-hand syndrome Slovenian type [MIM:610140]. Heart-hand syndrome (HHS) is a clinically and genetically heterogeneous disorder characterized by the co-occurrence of a congenital cardiac disease and limb malformations.,disease:Defects in LMNA are the cause of Hutchinson-Gilford progeria syndrome (HGPS) [MIM:176670]. HGPS is a rare genetic disorder characterized by features reminiscent of marked premature aging.,disease:Defects in LMNA are the cause of limb-girdle muscular dystrophy type 1B (LGMD1B) [MIM:159001]. LGMD1B is an autosomal dominant degenerative myopathy with age-related atrioventricular cardiac conduction disturbances, dilated cardiomyopathy, and the absence of early contractures. LGMD1B is characterized by slowly progressive skeletal muscle weakness of the hip and shoulder girdles. Muscle biopsy shows mild dystrophic changes.,disease:Defects in LMNA are the cause of mandibuloacral dysplasia with type A lipodystrophy (MADA) [MIM:248370]. Mandibuloacral dysplasia (MAD) is a rare autosomal recessive disorder characterized by mandibular and clavicular hypoplasia, acroosteolysis, delayed closure of the cranial suture, joint contractures, and types A or B patterns of lipodystrophy. Type A lipodystrophy observed in MADA, is characterized by fat loss restricted to the extremities.,function:Lamins are components of the nuclear lamina, a fibrous layer on the nucleoplasmic side of the inner nuclear membrane, which is thought to provide a framework for the nuclear envelope and may also interact with chromatin. Lamin A and C are present in equal amounts in the lamina of mammals.,miscellaneous:The structural integrity of the lamina is strictly controlled by the cell cycle, as seen by the disintegration and formation of the nuclear envelope in prophase and telophase, respectively.,miscellaneous:There are three types of lamins in human cells: A, B, and C.,PTM:Increased phosphorylation of the lamins occurs before envelope disintegration and probably plays a role in regulating lamin associations.,PTM:The C-terminal 18 residues are removed by proteolytic cleavage in isoform A. Proteolytic cleavage requires prior farnesylation and absence of farnesylation blocks cleavage.,similarity:Belongs to the intermediate filament family.,subunit:Homodimer of lamin A and lamin C. Interacts with lamin-associated polypeptides IA, IB and TMPO-alpha, RB1 and with emerin. Interacts with SREBF1, SREBF2 and TMEM43 (By similarity). Proteolytically processed isoform A interacts with NARF.,
- 功能:
- nucleus organization, nuclear envelope organization, gamete generation, spermatogenesis, muscle organ development, endomembrane organization, membrane organization, sexual reproduction, multicellular organism reproduction, male gamete generation, reproductive process in a multicellular organism,
- 细胞定位:
- Nucleus . Nucleus envelope . Nucleus lamina. Nucleus, nucleoplasm. Nucleus matrix . Farnesylation of prelamin-A/C facilitates nuclear envelope targeting and subsequent cleavage by ZMPSTE24/FACE1 to remove the farnesyl group produces mature lamin-A/C, which can then be inserted into the nuclear lamina. EMD is required for proper localization of non-farnesylated prelamin-A/C.; [Isoform C]: Nucleus speckle .
- 组织表达:
- In the arteries, prelamin-A/C accumulation is not observed in young healthy vessels but is prevalent in medial vascular smooth muscle cells (VSMCs) from aged individuals and in atherosclerotic lesions, where it often colocalizes with senescent and degenerate VSMCs. Prelamin-A/C expression increases with age and disease. In normal aging, the accumulation of prelamin-A/C is caused in part by the down-regulation of ZMPSTE24/FACE1 in response to oxidative stress.
- June 19-2018
- WESTERN IMMUNOBLOTTING PROTOCOL
- June 19-2018
- IMMUNOHISTOCHEMISTRY-PARAFFIN PROTOCOL
- June 19-2018
- IMMUNOFLUORESCENCE PROTOCOL
- September 08-2020
- FLOW-CYTOMEYRT-PROTOCOL
- May 20-2022
- Cell-Based ELISA│解您多样本WB检测之困扰
- July 13-2018
- CELL-BASED-ELISA-PROTOCOL-FOR-ACETYL-PROTEIN
- July 13-2018
- CELL-BASED-ELISA-PROTOCOL-FOR-PHOSPHO-PROTEIN
- July 13-2018
- Antibody-FAQs