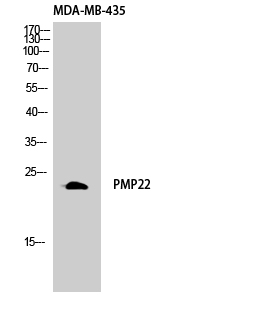
Total GAS3 Cell-Based Colorimetric ELISA Kit
- 货号:KA3362C
- 应用:ELISA
- 种属:Human;Mouse;Rat
- 其他名称:
- Peripheral myelin protein 22 (PMP-22) (Growth arrest-specific protein 3) (GAS-3)
- 背景:
- disease:Defects in PMP22 are a cause of Dejerine-Sottas syndrome (DSS) [MIM:145900]; also known as Dejerine-Sottas neuropathy (DSN) or hereditary motor and sensory neuropathy III (HMSN3). DSS is a severe degenerating neuropathy of the demyelinating Charcot-Marie-Tooth disease category, with onset by age 2 years. DSS is characterized by motor and sensory neuropathy with very slow nerve conduction velocities, increased cerebrospinal fluid protein concentrations, hypertrophic nerve changes, delayed age of walking as well as areflexia. There are both autosomal dominant and autosomal recessive forms of Dejerine-Sottas syndrome.,disease:Defects in PMP22 are a cause of hereditary neuropathy with liability to pressure palsies (HNPP) [MIM:162500]; an autosomal dominant disorder characterized by transient episodes of decreased perception or peripheral nerve palsies after slight traction, compression or minor traumas.,disease:Defects in PMP22 are the cause of Charcot-Marie-Tooth disease type 1A (CMT1A) [MIM:118220]; also known as hereditary motor and sensory neuropathy IA. CMT1A is a form of Charcot-Marie-Tooth disease, the most common inherited disorder of the peripheral nervous system. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathy or CMT1, and primary peripheral axonal neuropathy or CMT2. Neuropathies of the CMT1 group are characterized by severely reduced nerve conduction velocities (less than 38 m/sec), segmental demyelination and remyelination with onion bulb formations on nerve biopsy, slowly progressive distal muscle atrophy and weakness, absent deep tendon reflexes, and hollow feet. CMT1A inheritance is autosomal dominant.,disease:Defects in PMP22 are the cause of Charcot-Marie-Tooth disease type 1E (CMT1E) [MIM:118300]; also known as Charcot-Marie-Tooth disease and deafness autosomal dominant. CMT1E is an autosomal dominant form of Charcot-Marie-Tooth disease characterized by the association of sensorineural hearing loss with peripheral demyelinating neuropathy.,disease:Defects in PMP22 may be a cause of inflammatory demyelinating polyneuropathy (IDP) [MIM:139393]. IDP is a putative autoimmune disorder presenting in an acute (AIDP) or chronic form (CIDP). The acute form is also known as Guillain-Barre syndrome.,function:Might be involved in growth regulation, and in myelinization in the peripheral nervous system.,similarity:Belongs to the PMP-22/EMP/MP20 family.,
- 功能:
- regulation of action potential, cellular ion homeostasis, cell-cell signaling, synaptic transmission, ensheathment of neurons, peripheral nervous system development, negative regulation of cell proliferation, axon ensheathment,cellular component assembly involved in morphogenesis, transmission of nerve impulse, regulation of action potential in neuron, cellular homeostasis, myelin assembly, cellular component morphogenesis, regulation of cell proliferation,regulation of membrane potential, myelination, homeostatic process, chemical homeostasis, ion homeostasis,neurological system process, cellular chemical homeostasis,
- 细胞定位:
- Cell membrane ; Multi-pass membrane protein .