MT-ATP8 Polyclonal Antibody
- 货号:YT6262
- 应用:IHC;ELISA
- 种属:Human;Rat
- 简介:
- >>Oxidative phosphorylation;>>Metabolic pathways;>>Thermogenesis;>>Alzheimer disease;>>Parkinson disease;>>Amyotrophic lateral sclerosis;>>Huntington disease;>>Prion disease;>>Pathways of neurodegeneration - multiple diseases;>>Chemical carcinogenesis - reactive oxygen species;>>Diabetic cardiomyopathy
- 基因名称:
- MT-ATP8 ATP8 ATPASE8 MTATP8
- 免疫原:
- Synthesized peptide derived from human MT-ATP8 AA range: 30-110
- 特异性:
- This antibody detects endogenous levels of human MT-ATP8
- 组成:
- Liquid in PBS containing 50% glycerol, 0.5% BSA and 0.02% sodium azide.
- 来源:
- Polyclonal, Rabbit,IgG
- 稀释:
- IHC 1:50-200, ELISA(peptide)1:5000-20000
- 纯化工艺:
- The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen.
- 储存:
- -15°C to -25°C/1 year(Do not lower than -25°C)
- 其他名称:
- ATP synthase protein 8 (A6L;F-ATPase subunit 8)
- 背景:
- disease:Defects in MT-ATP6 are a cause of infantile bilateral striatal necrosis [MIM:500003]. Bilateral striatal necrosis is a neurological disorder resembling Leigh syndrome.,disease:Defects in MT-ATP6 are a cause of Leber hereditary optic neuropathy (LHON) [MIM:535000]. LHON is a maternally inherited disease resulting in acute or subacute loss of central vision, due to optic nerve dysfunction. Cardiac conduction defects and neurological defects have also been described in some patients. LHON results from primary mitochondrial DNA mutations affecting the respiratory chain complexes.,disease:Defects in MT-ATP6 are a cause of Leigh syndrome (LS) [MIM:256000]. LS is a severe neurological disorder characterized by bilaterally symmetrical necrotic lesions in subcortical brain regions.,disease:Defects in MT-ATP6 are the cause of neurogenic muscle weakness, ataxia, and retinitis pigmentosa (NARP) [MIM:551500].,disease:Defects in MT-CO3 are a cause of cytochrome c oxidase deficiency (COX deficiency) [MIM:220110]; also called mitochondrial complex IV deficiency. COX deficiency is a clinically heterogeneous disorder. The clinical features are ranging from isolated myopathy to severe multisystem disease, with onset from infancy to adulthood.,disease:Defects in MT-CO3 are a cause of Leber hereditary optic neuropathy (LHON) [MIM:535000]. LHON is a maternally inherited disease resulting in acute or subacute loss of central vision, due to optic nerve dysfunction. Cardiac conduction defects and neurological defects have also been described in some patients. LHON results from primary mitochondrial DNA mutations affecting the respiratory chain complexes.,disease:Defects in MT-CO3 are associated with recurrent myoglobinuria [MIM:550500]. Myoglobinuria consists of excretion of myoglobin in the urine.,disease:Defects in MT-CO3 are found in mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS) syndrome, a genetically heterogeneous disorder, characterized by episodic vomiting, seizures, and recurrent cerebral insults resembling strokes and causing hemiparesis, hemianopsia, or cortical blindness.,function:Mitochondrial membrane ATP synthase (F(1)F(0) ATP synthase or Complex V) produces ATP from ADP in the presence of a proton gradient across the membrane which is generated by electron transport complexes of the respiratory chain. F-type ATPases consist of two structural domains, F(1) - containing the extramembraneous catalytic core and F(0) - containing the membrane proton channel, linked together by a central stalk and a peripheral stalk. During catalysis, ATP synthesis in the catalytic domain of F(1) is coupled via a rotary mechanism of the central stalk subunits to proton translocation. Key component of the proton channel; it may play a direct role in the translocation of protons across the membrane.,function:Mitochondrial membrane ATP synthase (F(1)F(0) ATP synthase or Complex V) produces ATP from ADP in the presence of a proton gradient across the membrane which is generated by electron transport complexes of the respiratory chain. F-type ATPases consist of two structural domains, F(1) - containing the extramembraneous catalytic core and F(0) - containing the membrane proton channel, linked together by a central stalk and a peripheral stalk. During catalysis, ATP synthesis in the catalytic domain of F(1) is coupled via a rotary mechanism of the central stalk subunits to proton translocation. Part of the complex F(0) domain. Minor subunit located with subunit a in the membrane.,function:Subunits I, II and III form the functional core of the enzyme complex.,similarity:Belongs to the ATPase A chain family.,similarity:Belongs to the ATPase protein 8 family.,similarity:Belongs to the cytochrome c oxidase subunit 3 family.,subunit:F-type ATPases have 2 components, CF(1) - the catalytic core - and CF(0) - the membrane proton channel.,subunit:F-type ATPases have 2 components, CF(1) - the catalytic core - and CF(0) - the membrane proton channel. CF(1) has five subunits: alpha(3), beta(3), gamma(1), delta(1), epsilon(1). CF(0) has three main subunits: a, b and c.,
- 功能:
- disease:Defects in MT-ATP6 are a cause of infantile bilateral striatal necrosis [MIM:500003]. Bilateral striatal necrosis is a neurological disorder resembling Leigh syndrome.,disease:Defects in MT-ATP6 are a cause of Leber hereditary optic neuropathy (LHON) [MIM:535000]. LHON is a maternally inherited disease resulting in acute or subacute loss of central vision, due to optic nerve dysfunction. Cardiac conduction defects and neurological defects have also been described in some patients. LHON results from primary mitochondrial DNA mutations affecting the respiratory chain complexes.,disease:Defects in MT-ATP6 are a cause of Leigh syndrome (LS) [MIM:256000]. LS is a severe neurological disorder characterized by bilaterally symmetrical necrotic lesions in subcortical brain regions.,disease:Defects in MT-ATP6 are the cause of neurogenic muscle weakness, ataxia, and retinitis pigmentosa (NA
- 细胞定位:
- Mitochondrion membrane; Single-pass membrane protein.
- 组织表达:
- Blood,Bone fossil,Bones,Breast cancer,Distant normal tissue,Endometrial adenocarcin
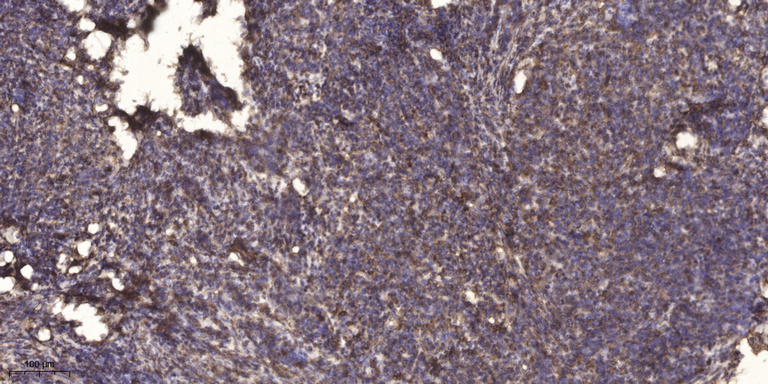
- Immunohistochemical analysis of paraffin-embedded human cervical carcinoma. 1, Antibody was diluted at 1:200(4° overnight). 2, Tris-EDTA,pH9.0 was used for antigen retrieval. 3,Secondary antibody was diluted at 1:200(room temperature, 45min).