ApoA-I Monoclonal Antibody
- 货号:YM0030
- 应用:WB;ELISA
- 种属:Human
- 简介:
- >>PPAR signaling pathway;>>Fat digestion and absorption;>>Vitamin digestion and absorption;>>Cholesterol metabolism;>>African trypanosomiasis;>>Lipid and atherosclerosis
- 免疫原:
- Purified recombinant fragment of human ApoA-I expressed in E. Coli.
- 特异性:
- ApoA-I Monoclonal Antibody detects endogenous levels of ApoA-I protein.
- 组成:
- Liquid in PBS containing 50% glycerol, 0.5% BSA and 0.02% sodium azide.
- 稀释:
- WB 1:500 - 1:2000. ELISA: 1:10000. Not yet tested in other applications.
- 纯化工艺:
- Affinity purification
- 储存:
- -15°C to -25°C/1 year(Do not lower than -25°C)
- 其他名称:
- APOA1;Apolipoprotein A-I;Apo-AI;ApoA-I;Apolipoprotein A1
- 背景:
- This gene encodes apolipoprotein A-I, which is the major protein component of high density lipoprotein (HDL) in plasma. The encoded preproprotein is proteolytically processed to generate the mature protein, which promotes cholesterol efflux from tissues to the liver for excretion, and is a cofactor for lecithin cholesterolacyltransferase (LCAT), an enzyme responsible for the formation of most plasma cholesteryl esters. This gene is closely linked with two other apolipoprotein genes on chromosome 11. Defects in this gene are associated with HDL deficiencies, including Tangier disease, and with systemic non-neuropathic amyloidosis. Alternative splicing results in multiple transcript variants, at least one of which encodes a preproprotein. [provided by RefSeq, Dec 2015],
- 功能:
- disease:Defects in APOA1 are a cause of amyloidosis type 8 (AMYL8) [MIM:105200]; also known as systemic non-neuropathic amyloidosis or Ostertag-type amyloidosis. AMYL8 is a hereditary generalized amyloidosis due to deposition of apolipoprotein A1, fibrinogen and lysozyme amyloids. Viscera are particularly affected. There is no involvement of the nervous system. Clinical features include renal amyloidosis resulting in nephrotic syndrome, arterial hypertension, hepatosplenomegaly, cholestasis, petechial skin rash.,disease:Defects in APOA1 are a cause of high density lipoprotein deficiency type 2 (HDLD2) [MIM:604091]; also known as familial hypoalphalipoproteinemia (FHA). Inheritance is autosomal dominant.,disease:Defects in APOA1 are a cause of the low HDL levels observed in high density lipoprotein deficiency type 1 (HDLD1) [MIM:205400]; also known as analphalipoproteinemia or Tangier dis
- 组织表达:
- Major protein of plasma HDL, also found in chylomicrons. Synthesized in the liver and small intestine. The oxidized form at Met-110 and Met-136 is increased in individuals with increased risk for coronary artery disease, such as in carrier of the eNOSa/b genotype and exposure to cigarette smoking. It is also present in increased levels in aortic lesions relative to native ApoA-I and increased levels are seen with increasing severity of disease.

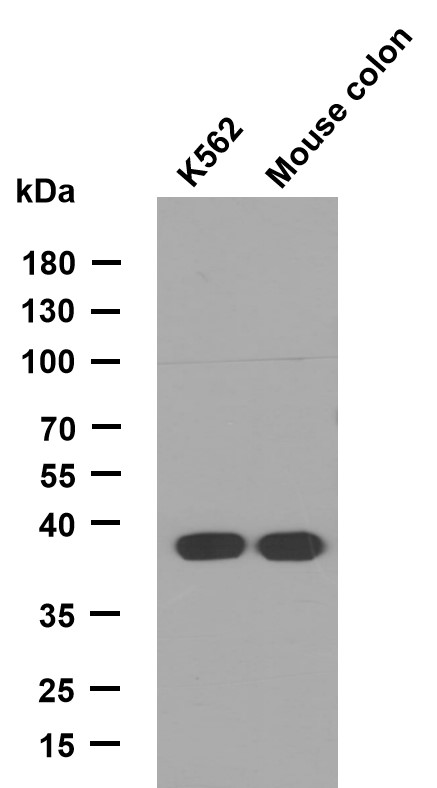
- Western Blot analysis using ApoA-I Monoclonal Antibody against HepG2 cell lysate (1) and human serum (2).