Sarcoglycan α Polyclonal Antibody
- Catalog No.:YT4215
- Applications:WB;IHC
- Reactivity:Human;Mouse
- Target:
- Sarcoglycan α
- Fields:
- >>Hypertrophic cardiomyopathy;>>Arrhythmogenic right ventricular cardiomyopathy;>>Dilated cardiomyopathy;>>Viral myocarditis
- Gene Name:
- SGCA
- Protein Name:
- Alpha-sarcoglycan
- Human Gene Id:
- 6442
- Human Swiss Prot No:
- Q16586
- Mouse Gene Id:
- 20391
- Mouse Swiss Prot No:
- P82350
- Immunogen:
- The antiserum was produced against synthesized peptide derived from human SGCA. AA range:161-210
- Specificity:
- Sarcoglycan α Polyclonal Antibody detects endogenous levels of Sarcoglycan α protein.
- Formulation:
- Liquid in PBS containing 50% glycerol, 0.5% BSA and 0.02% sodium azide.
- Source:
- Polyclonal, Rabbit,IgG
- Dilution:
- WB 1:500-2000;IHC 1:50-300
- Purification:
- The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen.
- Concentration:
- 1 mg/ml
- Storage Stability:
- -15°C to -25°C/1 year(Do not lower than -25°C)
- Other Name:
- SGCA;ADL;DAG2;Alpha-sarcoglycan;Alpha-SG;50 kDa dystrophin-associated glycoprotein;50DAG;Adhalin;Dystroglycan-2
- Observed Band(KD):
- 43kD
- Background:
- sarcoglycan alpha(SGCA) Homo sapiens This gene encodes a component of the dystrophin-glycoprotein complex (DGC), which is critical to the stability of muscle fiber membranes and to the linking of the actin cytoskeleton to the extracellular matrix. Its expression is thought to be restricted to striated muscle. Mutations in this gene result in type 2D autosomal recessive limb-girdle muscular dystrophy. Multiple transcript variants encoding different isoforms have been found for this gene. [provided by RefSeq, Oct 2008],
- Function:
- disease:Defects in SGCA are the cause of limb-girdle muscular dystrophy type 2D (LGMD2D) [MIM:608099]; also known as Duchenne-like muscular dystrophy autosomal recessive type 2 or severe childhood autosomal recessive muscular dystrophy (SCARMD). LGMD2D is an autosomal recessive degenerative myopathy characterized by progressive muscle wasting from early childhood with loss of independent ambulation by teenage years. Muscle biopsy shows necrosis, decreased immunostaining for alpha sarcoglycan, and adhalin deficiency. The phenotype is less severe than LGMD2C.,function:Component of the sarcoglycan complex, a subcomplex of the dystrophin-glycoprotein complex which forms a link between the F-actin cytoskeleton and the extracellular matrix.,online information:SGCA mutations in LGMD2D,similarity:Belongs to the sarcoglycan alpha/epsilon family.,subunit:Interacts with the syntrophin SNTA1. Cross-
- Subcellular Location:
- Cell membrane, sarcolemma ; Single-pass type I membrane protein . Cytoplasm, cytoskeleton .
- Expression:
- Most strongly expressed in skeletal muscle. Also expressed in cardiac muscle and, at much lower levels, in lung. In the fetus, most abundant in cardiac muscle and, at lower levels, in lung. Also detected in liver and kidney. Not expressed in brain.
Sacubitril/Valsartan Alleviates Experimental Autoimmune Myocarditis by Inhibiting Th17 Cell Differentiation Independently of the NLRP3 Inflammasome Pathway. Frontiers in Pharmacology Front Pharmacol. 2021; 12: 727838 WB Mouse 1:1000 Myocardial tissues
- June 19-2018
- WESTERN IMMUNOBLOTTING PROTOCOL
- June 19-2018
- IMMUNOHISTOCHEMISTRY-PARAFFIN PROTOCOL
- June 19-2018
- IMMUNOFLUORESCENCE PROTOCOL
- September 08-2020
- FLOW-CYTOMEYRT-PROTOCOL
- May 20-2022
- Cell-Based ELISA│解您多样本WB检测之困扰
- July 13-2018
- CELL-BASED-ELISA-PROTOCOL-FOR-ACETYL-PROTEIN
- July 13-2018
- CELL-BASED-ELISA-PROTOCOL-FOR-PHOSPHO-PROTEIN
- July 13-2018
- Antibody-FAQs
- Products Images
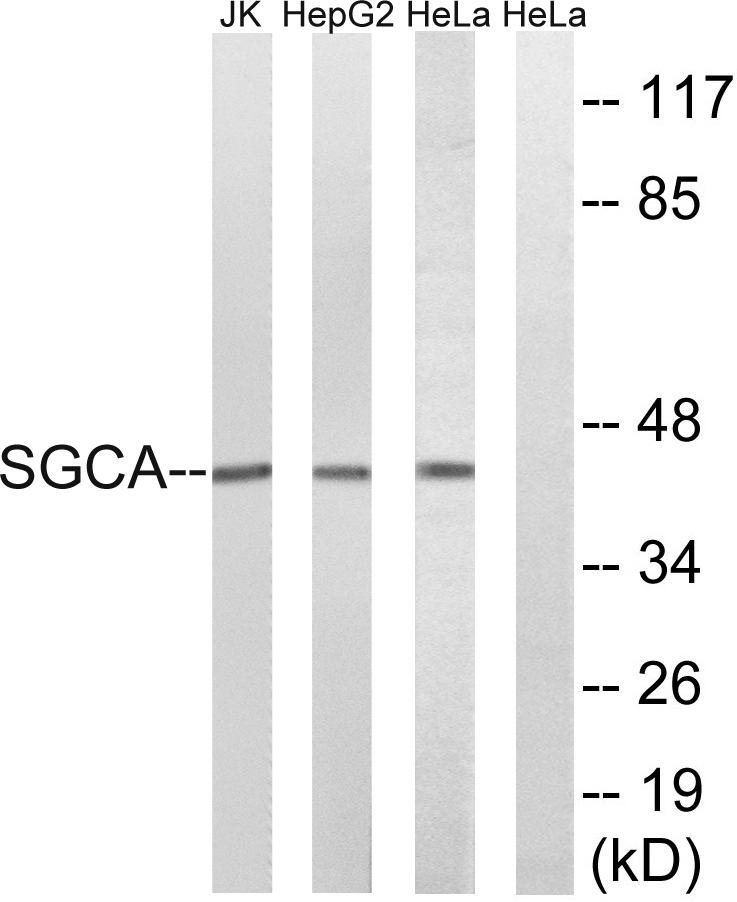
- Western blot analysis of lysates from HeLa, HepG2, and Jurkat cells, using SGCA Antibody. The lane on the right is blocked with the synthesized peptide.
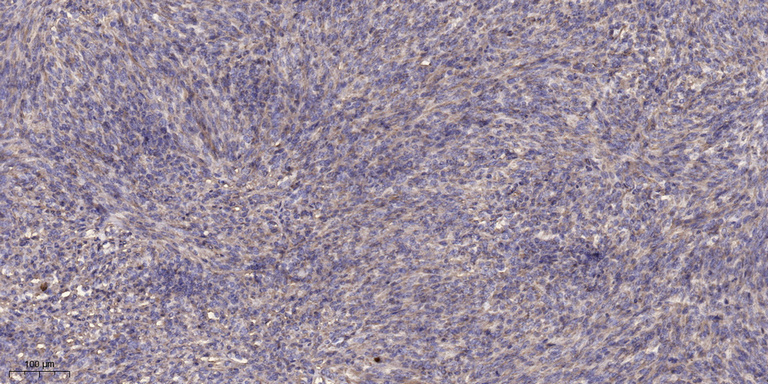
- Immunohistochemical analysis of paraffin-embedded human Colon cancer. 1, Antibody was diluted at 1:200(4° overnight). 2, Tris-EDTA,pH9.0 was used for antigen retrieval. 3,Secondary antibody was diluted at 1:200(room temperature, 45min).